

On Nov 25, 2020, the National Medical Products Administration (NMPA) and seven other ministries released a work plan to promote the innovative development of medical product regulation in the Guangdong-Hong Kong-Macao Greater Bay Area. The plan aims to establish an effective interface between the Chinese mainland’s regulatory system and those of Hong Kong and Macao. Given Hong Kong’s strengths in innovation and technology, especially in biomedical fields, the plan presents valuable opportunities to Hong Kong. However, Hong Kong lacks an independent regulatory body for medical product review and approval, hindering its competitiveness in promoting international innovation. The recommendation is to expedite the incorporation of Hong Kong’s self-developed devices into the mainland’s review and approval system, facilitating the industrialization of Hong Kong’s biotechnology and the regulatory harmonization between the two places, thereby accelerating the approval and surveillance capacity in both the mainland and Hong Kong, which is of great significance.
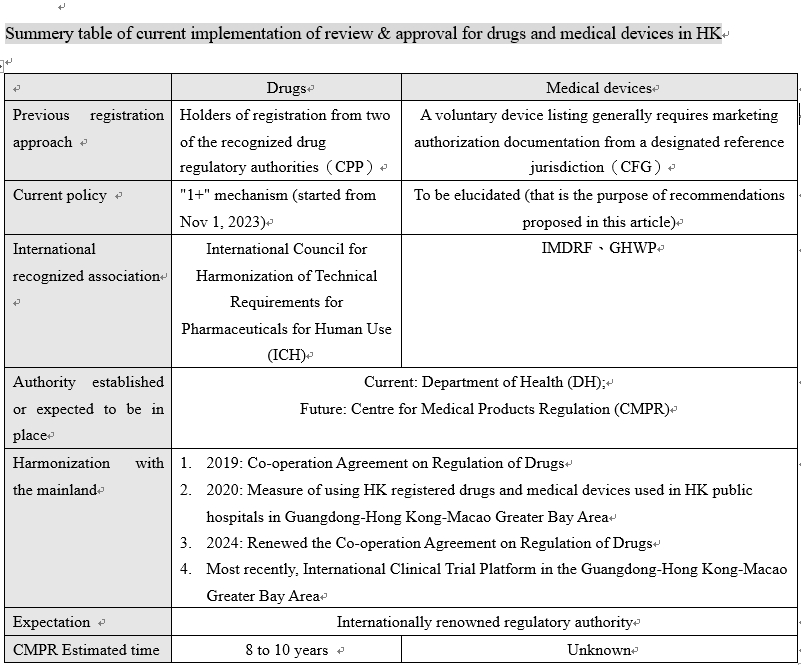
The existing regulatory framework and current progress for relevant authority in HK
Foster establishment of review and approval for medical devices in Hong Kong; the challenge on mainland: The mechanism for approving drugs has been initiated, but for medical devices, particularly Class III medical devices, has not yet been clarified.
The implementation of the “Measure of using HK registered drugs and medical devices used in Hong Kong public hospitals in Guangdong-Hong Kong-Macao Greater Bay Area” has lasted for more than three years
READ MORE: HKUST to establish city's third medical school by mid-2027
(1) Hong Kong research organizations face the same dilemma in translating their products to the market in registration and approval. The 2023 Policy Address proposed to establish a “Tier 1” medical product review and approval authority in Hong Kong, implementing the “1+” mechanism: application for drug registration can be made through the issuance of a Certificate of Pharmaceutical Product (CPP) from a country or region, local data and expert reports. However, the approval mechanism for devices is not specified.
(2) The perception of medical device development (researched in Hong Kong and manufactured on the mainland) does not reflect the practical conversion process. For Class III implantable devices, research bodies typically need to establish a company on the mainland, apply for and obtain the medical device registration certificate. University research projects require the university to hold the patent rights, apply for patents and licensing. The mainland’s larger clinical sample size and the market size also makes it the preferred location for conducting clinical trials, especially for innovative Class III devices.
Medical Device Administrative Control System (MDACS): Voluntary List without Registration and Approval
(3) Hong Kong‘s Medical Device Administrative Control System (MDACS) managed by the Medical Device Division (MDD) under the Department of Health (DH) includes a voluntary medical device listing system and a post-marketing surveillance reporting mechanism, but no specific regulations have been enacted yet. Device classification and compliance with safety, quality, and technical standards are required for hospital procurement. The system is intended to facilitate a smooth transition to a future statutory regulatory framework aligned with international best practices.
Medical Device Administrative Control System (MDACS): Voluntary List without Registration and Approval
(4) Devices are classified into risk classes I to IV under the Medical Device Administrative Control System (MDACS). If a device is required to be listed, e.g., for hospital procurement, it must comply with safety and efficacy principles. The company must provide quality management system certificates (e.g., ISO 13485), device manuals, technical standards compliance, and sales approval documents (Certificate for Foreign Government, CFG). After being granted market access, the device is subject to post-market surveillance.
The mainland policy on Guangdong, Hong Kong and Macao and the measure on “interoperability of devices” for Hong Kong can address the urgent needs; however, it is not sufficient to support domestic innovation, research and development.
Given the current situation of the devices exchanged between Hong Kong and the mainland side of the GBA, this is only an extension of the use of drugs and devices already in use in Hong Kong to the mainland side of the GBA. The implementation of the “Measure of using HK registered drugs and medical devices used in Hong Kong public hospitals in Guangdong-Hong Kong-Macao Greater Bay Area” has lasted for more than three years, and as of Jan 16, 2024, 28 types of medical devices have obtained permission to be used in 19 designated medical institutions, benefiting 4,509 patients. Worth mentioning, those already in use in Hong Kong were imported with approval from other markets notably the United States, European Union and Japan, which implies that a “precedent” is to be followed instead of mutual recognition that does not involve approval. This “interoperability” approach is not the same as supporting scientific research and clinical translation of medical product innovations. The purpose of “interoperability” is to address urgent clinical needs, so that medical institutions can directly use medical products imported for use in Hong Kong, and the functionality is unlike the conventional approval system.
Recommendations
Hong Kong has advantages in developing an independent review and approval system, either on its own or in collaboration with the mainland side of the GBA. With robust scientific research capacity alongside world top-ranking universities and medical schools as well as many international accreditation test laboratories as the foundation for review, the creation of an independent regulatory body for registration and approval is feasible, to be facilitated by internationalization, digitization, and a recognized regulatory system. Meanwhile, integration into the NMPA review and approval system should be planned in advance, with the following recommendations:
(1) Establishment and amendment of related laws: Improve Hong Kong’s medical device regulatory system, and formulate administrative regulations as the foundation, such as a Hong Kong Medical Devices Regulation Act. Hong Kong should formulate related departmental rules and regulations, so there is a sophisticated set of complementary measures to establish an end-to-end product life cycle management from premarket approval to post market surveillance.
(2) Review capability: It is essential to establish a talent pool with multidisciplinary expertise for review and assessment. In Hong Kong, universities and hospitals are closely connected, and the training of talent in interdisciplinary fields is a matter of great urgency, and resources are required. In addition, the Department of Health itself has a mechanism to assess the relevant testing organizations. In the future, it is necessary to expand its inspection capacity in the GBA in collaboration with the GBA branches of the NMPA.
(3) Clinical trials: Medical device sample requirement is generally 100 to 200 cases per arm, which is feasible in Hong Kong. Consideration can be given to the dual review system of clinical trials in the two jurisdictions, i.e., when clinical trials are reviewed by the Hong Kong authorities, they are also subject to review and approval by the NMPA. GBA has been facilitating the sharing of talent, data and samples, which established foundation for this mechanism.
In the future, when registering in Hong Kong, the registration certificate can be directly applied to the whole GBA through the form of “first approval and then verification” (conditional approval). Subsequent registration through a fast track designation by the NMPA will reduce the review time. Real-world data can support the review process. Within the corresponding predictable review time, the registration can then be converted into a regular registration certificate in a few years with the support of the relevant data, thus achieving a dual-review, dual-approval system.
READ MORE: UK, mainland top sources of HK’s non-locally trained doctors
(4) Mutual recognition of devices with the NMPA: As there are many categories of medical devices, it is inevitable that not all of them can achieve the mutual recognition at the beginning, it is recommended that priority should be given to commonly used and matured devices of low risk and with large quantities, for instance, orthopedic devices or implants.
(5) International endorsement: Three main steps: 1) In addition to mutual recognition with the mainland, it is more important to engage in international activities through bilateral and multilateral communication channels. 2) The capability to join international organizations is essential for a regulatory body to be recognized. 3) If Hong Kong becomes an international regulatory body, it can leverage World Health Organization accreditation.
(6) Insurance/Medicare system: A set of rational device pricing and administration mechanism, or leave it to the market to decide whether the voluntary health insurance and commercial insurance will prevail, are the key issues that should be reconsidered.
Hsueh Yu-sheng is from the Department of Orthopaedics and Traumatology, the Chinese University of Hong Kong.
Qin Ling is from the CUHK Hong Kong-Shenzhen Innovation and Technology Institute (Futian).
The views do not necessarily reflect those of China Daily.